Addition Reactions of Alkynes
Addition Reactions of Alkynes
– Many of the reactions of alkynes are similar to the corresponding reactions of alkenes because both involve pi bonds between two carbon atoms.
– Like the pi bond of an alkene, the pi bonds of an alkyne are electron-rich, and they readily undergo addition reactions.
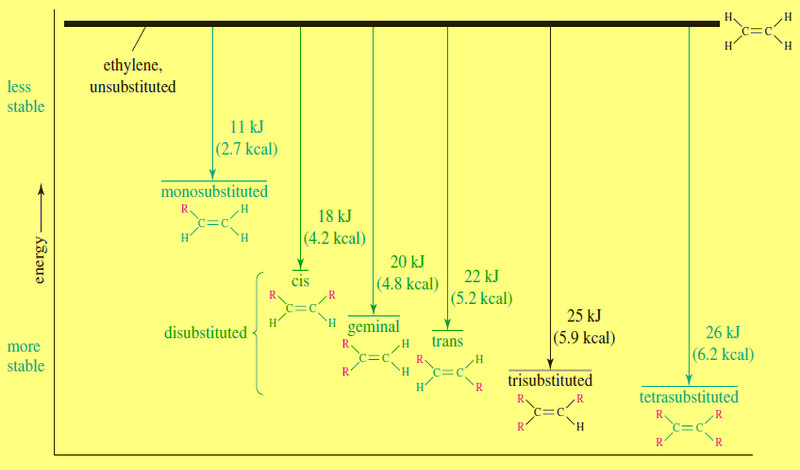
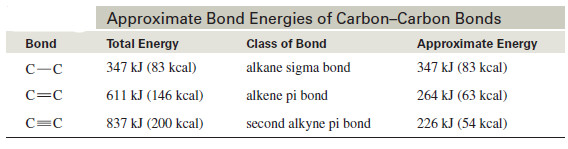
– The following table shows how the energy differences between the kinds of carbon–carbon bonds can be used to estimate how much energy it takes to break a particular bond.
– The bond energy of the alkyne triple bond is only about 226 kJ (54 kcal) more than the bond energy of an alkene double bond.
– This is the energy needed to break one of the pi bonds of an alkyne.
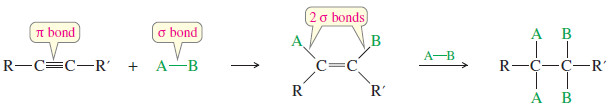
– Reagents add across the triple bonds of alkynes just as they add across the double bonds of alkenes.
– In effect, this reaction converts a pi bond of the alkyne and a sigma bond of the reagent into two new sigma bonds.
– Since sigma bonds are generally stronger than pi bonds, the reaction is usually exothermic.
– Alkynes have two pi bonds, so up to two molecules can add across the triple bond, depending on the reagents and the conditions
– We must consider the possibility of a double addition whenever a reagent adds across the triple bond of an alkyne.
– Some conditions may allow the reaction to stop after a single addition, while other conditions give double addition.
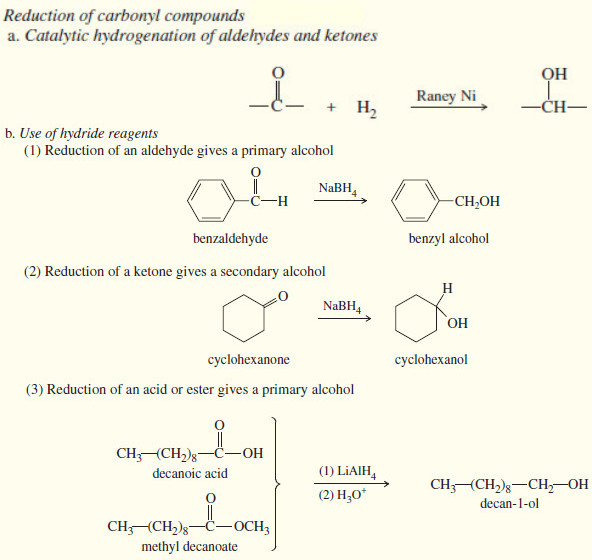
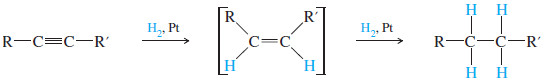
(1) Catalytic Hydrogenation alkynes to Alkanes
– In the presence of a suitable catalyst, hydrogen adds to an alkyne, reducing it to an alkane.
– For example, when either of the butyne isomers reacts with hydrogen and a platinum catalyst, the product is n-butane.
– Platinum, palladium, and nickel catalysts are commonly used in this reduction.
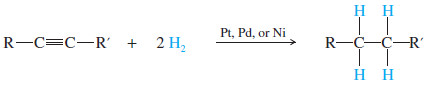
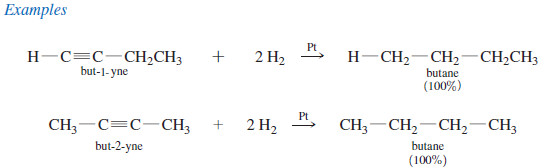
– Catalytic hydrogenation takes place in two stages, with an alkene intermediate.
– With efficient catalysts such as platinum, palladium, or nickel, it is usually impossible to stop the reaction at the alkene stage
(2) Catalytic Hydrogenation of alkynes to cis Alkenes
– Hydrogenation of an alkyne can be stopped at the alkene stage by using a “poisoned” (partially deactivated) catalyst made by treating a good catalyst with a compound that makes the catalyst less effective.
– Lindlar’s catalyst is a poisoned palladium catalyst, composed of powdered barium sulfate coated with palladium, poisoned with quinoline.
– Nickel boride (Ni2B) is a newer alternative to Lindlar’s catalyst that is more easily made and often gives better yields.
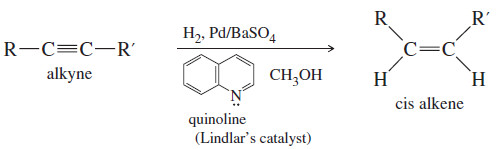
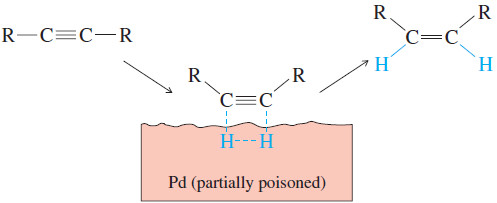
– The catalytic hydrogenation of alkynes is similar to the hydrogenation of alkenes, and both proceed with syn stereochemistry.
– In catalytic hydrogenation, the face of a pi bond contacts the solid catalyst, and the catalyst weakens the pi bond, allowing two hydrogen atoms to add .
– This simultaneous (or nearly simultaneous) addition of two hydrogen atoms on the same face of the alkyne ensures syn stereochemistry.
– In an internal alkyne, syn addition gives a cis product.
– For example, when hex-2-yne is hydrogenated using Lindlar’s catalyst, the product is cis-hex-2-ene.
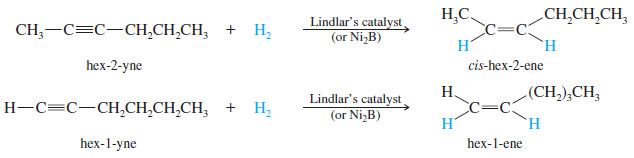
(3) Metal–Ammonia Reduction to trans Alkenes
– To form a trans alkene, two hydrogens must be added to the alkyne with anti stereochemistry.
– Sodium metal in liquid ammonia reduces alkynes with anti stereochemistry, so this reduction is used to convert alkynes to trans alkenes.
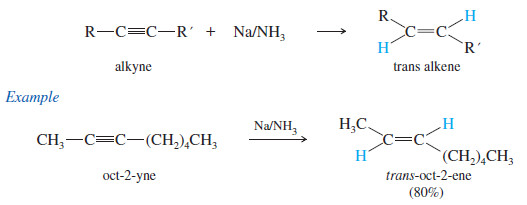
– Ammonia (bp -33Co ) is a gas at room temperature, but it is kept liquid by using dry ice to cool the reaction vessel.
– As sodium dissolves in liquid ammonia, it gives up electrons, which produce a deep blue color. It is these solvated electrons that actually reduce the alkyne.
![]()
– The metal–ammonia reduction proceeds by addition of an electron to the alkyne to form a radical anion, followed by protonation to give a neutral radical.
– Protons are provided by the ammonia solvent or by an alcohol added as a cosolvent.
– Addition of another electron, followed by another proton, gives the product
Meachanism: Metal–Ammonia Reduction of an Alkyne
– This mechanism involves addition of an electron, followed by a proton, then addition of a second electron, followed by a second proton.
Step 1: An electron adds to the alkyne, forming a radical anion
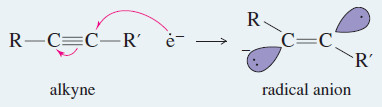
Step 2: The radical anion is protonated to give a radical.
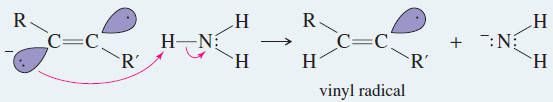
Step3 : An electron adds to the radical, forming an anion
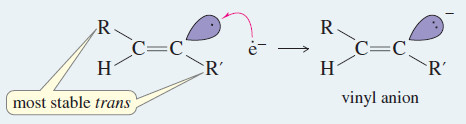
Step 4: Protonation of the anion gives an alkene.
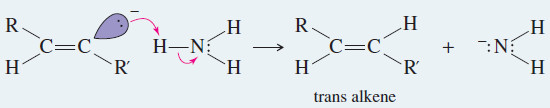
– The anti stereochemistry of the sodium–ammonia reduction appears to result from the greater stability of the vinyl radical in the trans configuration, where the alkyl groups are farther apart.
– An electron is added to the trans radical to give a trans vinyl anion, which is quickly protonated to the trans alkene.
(4) Addition of Halogens
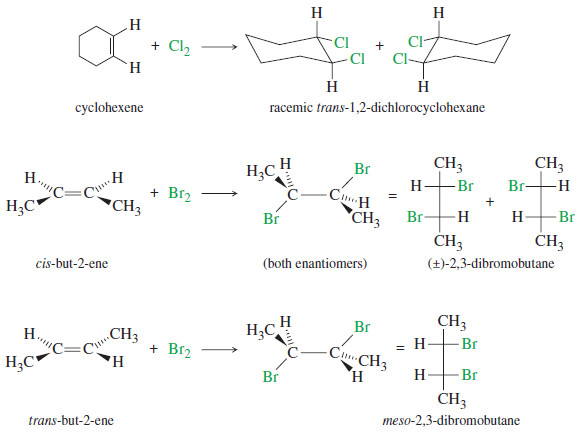
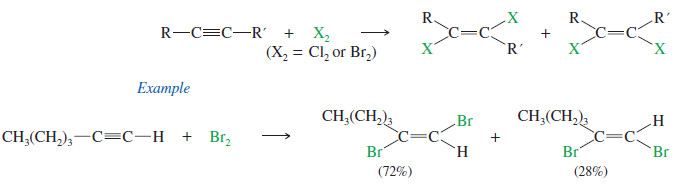
– Bromine and chlorine add to alkynes just as they add to alkenes.
– If 1 mole of halogen adds to 1 mole of an alkyne, the product is a dihaloalkene.
– The stereochemistry of addition may be either syn or anti, and the products are often mixtures of cis and trans isomers.
– If 2 moles of halogen add to 1 mole of an alkyne, a tetrahalide results.
– Sometimes it is difficult to keep the reaction from proceeding all the way to the tetrahalide even when we want it to stop at the dihalide
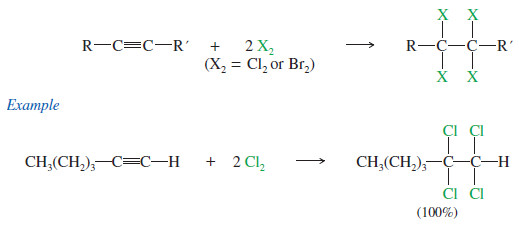
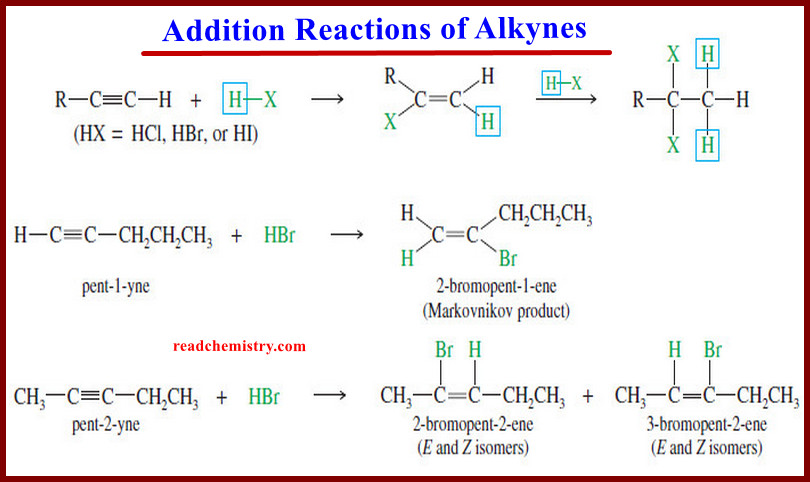
(5) Addition of Hydrogen Halides
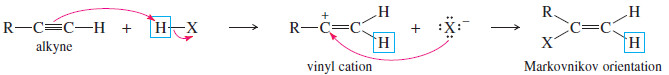
– Hydrogen halides add across the triple bond of an alkyne in much the same way they add across the alkene double bond.
– The initial product is a vinyl halide. When a hydrogen halide adds to a terminal alkyne, the product has the orientation predicted by Markovnikov’s rule.
– A second molecule of HX can add, usually with the same orientation as the first.
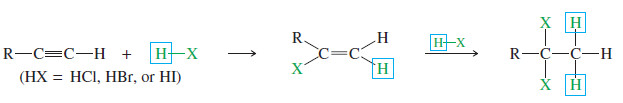
– For example, the reaction of pent-1-yne with HBr gives the Markovnikov product.
– In an internal alkyne such as pent-2-yne, however, the acetylenic carbon atoms are equally substituted, and a mixture of products results.
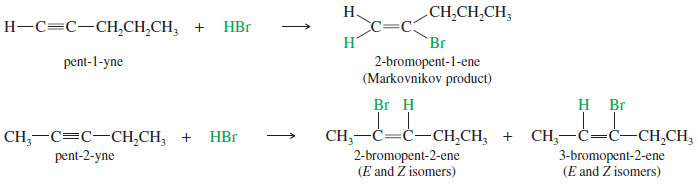
– The mechanism is similar to the mechanism of hydrogen halide addition to alkenes.
– The vinyl cation formed in the first step is more stable with the positive charge on the more highly substituted carbon atom. Attack by halide ion completes the reaction.
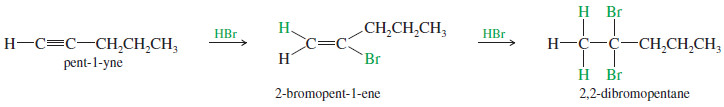
– When 2 moles of a hydrogen halide add to an alkyne, the second mole usually adds with the same orientation as the first.
– This consistent orientation leads to a geminal dihalide.
– For example, a double Markovnikov addition of HBr to pent-1-yne gives 2,2-dibromopentane
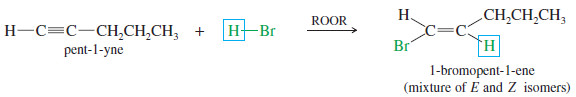
– In our subjects before , we saw the effect of peroxides on the addition of HBr to alkenes.
– Peroxides catalyze a free-radical chain reaction that adds HBr across the double bond of an alkene in the anti-Markovnikov sense.
– A similar reaction occurs with alkynes, with HBr adding with anti-Markovnikov orientation.
(6) Hydration of Alkynes to Ketones and Aldehydes
(A) Mercuric Ion-Catalyzed Hydration
– Alkynes undergo acid-catalyzed addition of water across the triple bond in the presence of mercuric ion as a catalyst.
– A mixture of mercuric sulfate in aqueous sulfuric acid is commonly used as the reagent.
– The hydration of alkynes is similar to the hydration of alkenes, and it also goes with Markovnikov orientation.
– The products are not the alcohols we might expect, however.
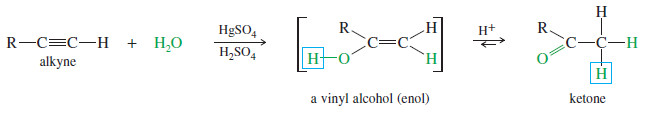
– Electrophilic addition of mercuric ion gives a vinyl cation, which reacts with water and loses a proton to give an organomercurial alcohol
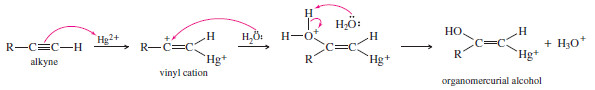
– Under the acidic reaction conditions, mercury is replaced by hydrogen to give a vinyl alcohol, called an enol
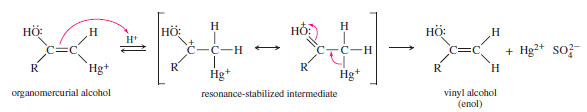
– Enols tend to be unstable and isomerize to the ketone form. As shown next, this isomerization involves the shift of a proton and a double bond.
– The (boxed) hydroxyl proton is lost, and a proton is regained at the methyl position, while the pi bond shifts from the (C=C) position to the (C=O) position. This type of rapid equilibrium is called a tautomerism.
– The one shown is the keto–enol tautomerism, which is covered in more detail Next. The keto form usually predominates.
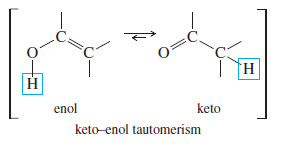
– In acidic solution, the keto–enol tautomerism takes place by addition of a proton to the adjacent carbon atom, followed by loss of the hydroxyl proton from oxygen
Meachanism: Acid-Catalyzed Keto–Enol Tautomerism
– Under acidic conditions, the proton first adds at its new position on the adjacent carbon atom, and then is removed from its old position in the hydroxyl group.
Step 1: Addition of a proton at the methylene group.
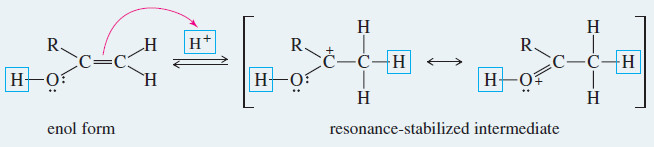
Step 2: Loss of the hydroxyl proton.
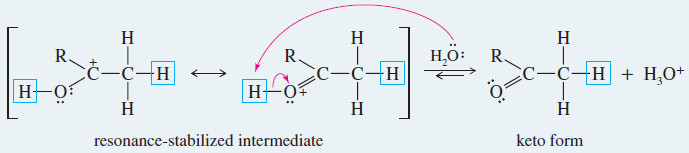
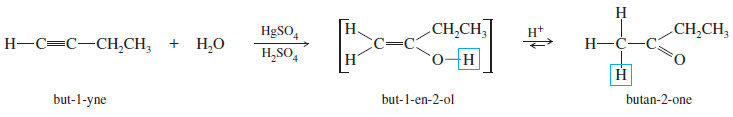
– For example, the mercuric-catalyzed hydration of but-1-yne gives but-1-en-2-ol as an intermediate.
– In the acidic solution, the intermediate quickly equilibrates to its more stable keto tautomer, butan-2-one.
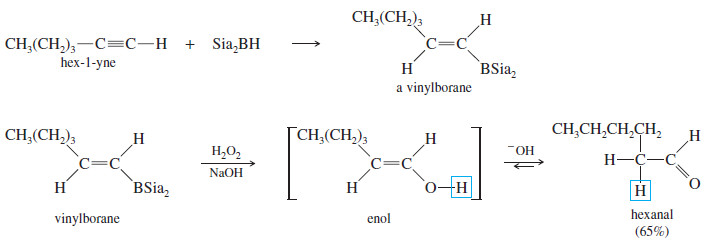
(B) Hydroboration–Oxidation
– Hydroboration–oxidation adds water across the double bonds of alkenes with anti-Markovnikov orientation.
– A similar reaction takes place with alkynes, except that a hindered dialkylborane must be used to prevent addition of two molecules of borane across the triple bond.
– Di(secondary isoamyl)borane, called “disiamylborane,” adds to the triple bond only once to give a vinylborane. (Amyl is an older common name for pentyl.)
– In a terminal alkyne, the boron atom bonds to the terminal carbon atom.
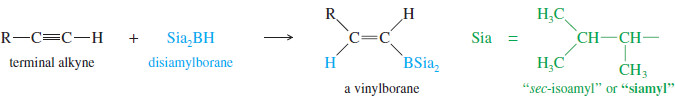
– Oxidation of the vinylborane (using basic hydrogen peroxide) gives a vinyl alcohol (enol), resulting from anti-Markovnikov addition of water across the triple bond.
– This enol quickly tautomerizes to its more stable carbonyl (keto) form.
– In the case of a terminal alkyne, the keto product is an aldehyde. This sequence is an excellent method for converting terminal alkynes to aldehydes.
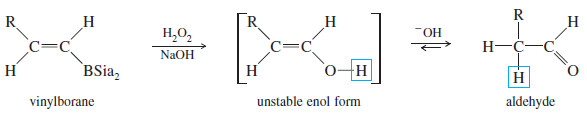
– Under basic conditions, the keto–enol tautomerism operates by a different mechanism than it does in acid.
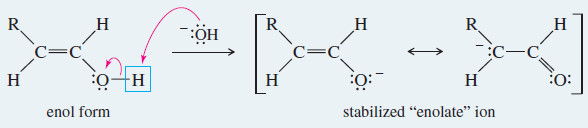
– In base, the proton is first removed from its old position in the OH group, and then replaced on carbon.
– In acid, the proton was first added on carbon, and then removed from the hydroxyl group.
Meachanism: Base-Catalyzed Keto–Enol Tautomerism
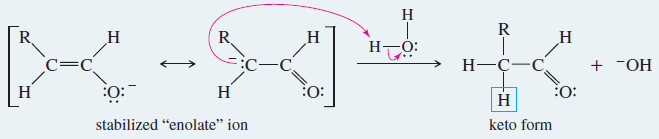
Under basic conditions, the proton is first removed from its old position in the enol, and then replaced in its new position on the adjacent carbon atom of the ketone or aldehyde.
Step 1: Loss of the hydroxyl proton.
Step 2: Reprotonation on the adjacent carbon atom.
– Hydroboration of hex-1-yne, for example, gives the vinylborane with boron on the less highly substituted carbon.
– Oxidation of this intermediate gives an enol that quickly tautomerizes to hexanal