Gibbs Helmholtz Equations

Gibbs Helmholtz Equations
– These are two equations derived by J.W. Gibbs and H.Von Helmholtz and are known as Gibbs Helmholtz equations.
– One equation can be expressed in terms of changes in free energy (ΔG) and enthalpy (ΔH), while the other can be expressed in terms of changes in internal energy (ΔE) and work function (ΔA).
– The former is generally employed and is applicable to all processes, chemical or physical, but in a closed system.
(a) In Terms of Free Energy and Enthalpy
– We know that:
dG = VdP – SdT
– At constant pressure dP = 0, then
dG = – SdT
– Let G1 be the initial free energy at temperature T, G1 + dG1, be the free energy at temperature T + dT.
Then
dG1 = – S1 dT …(i)
Where S1 is the entropy of the system in the initial state.
– Now suppose the free energy of the system in the final state is G2 at temperature T, and G2 + dG2 is the free energy at temperature T + dT in the final state.
Then
dG2 = – S2 dT …(ii)
where S2 is the entropy of the system in the final state.
– Subtracting (i) from (ii) we get:
dG2 – dG1 = –(S2 – S1) dT
d (ΔG) = – ΔS dT
– At constant pressure:
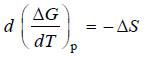 (iii)
(iii)
– We know
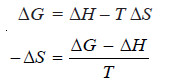 (iv)
(iv)
– Comparing equations (iii) and (iv):
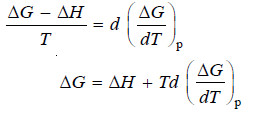
– This equation is called Gibbs Helmholtz equations in terms of free energy and enthalpy change at constant pressure.
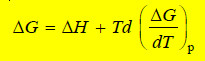
(b) In terms of Internal Energy and Work Function
– By definition the work function:
A = E – TS …(i)
or
ΔA = ΔE – TΔS
or
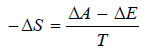
– Differentiating equation (i) we get:
dA = dE – TdS – SdT …(iii)
– From the first law of thermodynamics
dq = dE + PdV
and at constant volume dV = 0
dq = dE
– For a reversible change
![]()
or dq = TdS – dE …(iv)
– Comparing equations (iii) and (iv) we get:
dA = – SdT
– Let A1 be the work function in its initial state at temperature T and A1 + dA1 be the work function in its initial state at T + dT. And A2 be the work function in its final state at temperature T and A2 + dA2 be the work function in its final state at T + dT. Then
dA1 = – S1 dT …(v)
and
dA2 = – S2 dT …(vi)
where S1 and S2 are the entropies of the system in initial and final states of the system respectively.
– Subtracting equation (v) from equation (vi) we get
dA2 – dA1 = – (S2 – S1) dT
or
d (ΔA) = – ΔS dT
– At constant volume
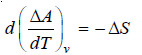
– From equation (ii) we have
![]()
– On comparison we have
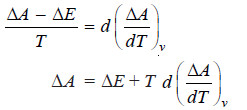
– This equation is called Gibbs Helmholtz equation in terms of internal energy and work function at
constant volume.
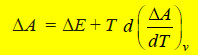
Importance of Gibbs Helmholtz Equations
– Gibb’s Helmholtz equations are used to calculate the heats of reaction (ΔH or ΔE) when ΔG or ΔA at two temperatures are given.
– This point is made clear in the following examples:
Solved Problems Gibbs Helmholtz Equations
Problem (1): For the reaction:
![]()
The values of enthalpy change and free energy change are – 68.32 and – 56.69 kcals respectively. Calculate the value of free energy change at 25ºC.
Solution:
– Using the Gibb’s Helmholtz equation:
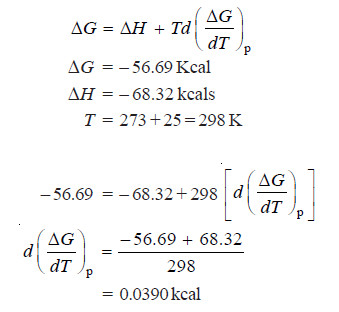
– Assuming that 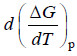 remains constant over this temperature range.
remains constant over this temperature range.
– At 30ºC we can write:
ΔG = 68.32 + 303 × 0.039
= – 68.32 + 11.817
= – 56.503 kcals
Problem (2): For the following reaction
N2 (g) + 3H2 (g) → 2NH3 (g)
The free energy changes at 25ºC and 35ºC are – 33.089 and – 28.018 kJ respectively. Calculate the heat of reaction.
Solution:
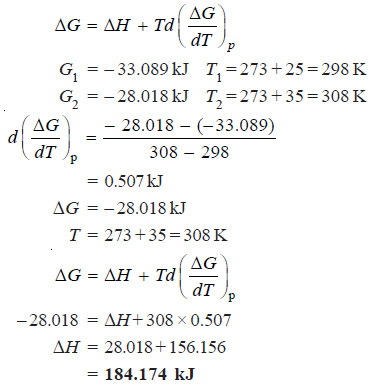
Conditions of Equilibrium and Criterion for a Spontaneous Process
(a) In Terms of Entropy Change
– The entropy of a system remains unchanged in a reversible change while it increases in an irreversible change i.e.
– Σ dS = 0 For Reversible change
– and Σ dS > 0 For Irreversible change
– For a system with its surroundings we can write
dSsystem + dSsurroundings = 0
and
dSsystem + dSsurroundings > 0
– combining the two relations, we have
dSsystem + dSsurroundings ≥ 0 …(i)
– If we assume the change in surroundings as reversible and surroundings evolve dq heat reversibly, then
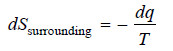
– From the first law of Thermodynamics
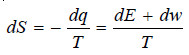
where dE is the increase in internal energy of the system and dw be the work done by the system.
– From equation (i) we get:
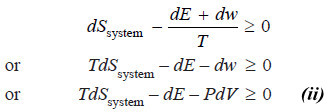
![]()
Case I. When E and V are constant
– In this case dE = 0 and dV = 0 the equation (i) reduces to
dSE.V ≥ 0
or
dSE.V > 0 for an irreversible change (spontaneous)
and
dSE.V = 0 for a reversible change (equilibrium)
Case II. When S and V are constant
– Here dS = 0 and dV = 0
– The equation (i) reduces to
– dE ≥ 0
or
– dE > 0 for an irreversible change (spontaneous)
and
– dE = 0 for a reversible change (equilibrium)
(b) In Terms of Enthalpy Change
– We know
H = E + PV
– on differentiating
dH = dE + PdV + VdP
or
– dE – PdV = – dH + VdP
– Putting in equation (ii)
TdS – dH + VdP ≥ 0
or
dH – VdP – TdS ≤ 0
At constant S and P
Here
dS = 0 and dP = 0
∴ we have dH ≤ 0
or
dH < 0 for an irreversible change (spontaneous)
and
dH = 0 for a reversible change (equilibrium)
(c) In Terms of Free Energy Change
– We know
G = H + TS
or
G = E + PV + TS
[∵ H = E + PV]
– on differentiating we get
dG = dE + PdV + VdP + TdS + SdT
TdS – dE – PdV = – dG + VdP – SdT
– Substituting in equations (ii) we get
– dG + VdP – SdT ≥ 0
or
dG – VdP + SdT ≤ 0
– At constant pressure in an isothermal process (T is also constant) this equation reduces to
dG ≤ 0
or
dG < 0 for an irreversible change (spontaneous)
and
dG = 0 for a reversible change (equilibrium)
– Thus the conditions for spontaneity and equilibrium may be summed up in the following Table: