
Precipitation Gravimetry
– In this topic, we will discuss the Precipitation Gravimetry as an important one of Gravimetric Analysis in analytical chemistry.
What is Gravimetric Analysis?
– Gravimetric analysis is a method to determine the quantity of an analyte based on the mass of a solid.
– Gravimetric methods of analysis are based on mass measurements with an analytical balance, an instrument that yields highly accurate and precise data.
– In fact, if you perform a gravimetric determination in your laboratory, you may make some of the most accurate and precise measurements of your life
Types of Gravimetric Analysis
– Gravimetric methods are quantitative methods that are based on determining the mass of a pure compound to which the analyte is chemically related.
– Several analytical methods are based on mass measurements as follow:
(1) precipitation gravimetry
– In precipitation gravimetry, the analyte is separated from a solution of the sample as a precipitate and is converted to a compound of known composition that can be weighed.
(2) volatilization gravimetry
– In volatilization gravimetry, the analyte is separated from other constituents of a sample by converting it to a gas of known chemical composition.
– The mass of the gas then serves as a measure of the analyte concentration.
(3) electrogravimetry
– In electrogravimetry, the analyte is separated by deposition on an electrode by an electrical current.
– The mass of this product then provides a measure of the analyte concentration.
(4) gravimetric titrimetry
– In gravimetric titrimetry, the mass of a reagent of known concentration required to react completely with the analyte provides the information needed to determine the analyte concentration.
(5) Atomic mass spectrometry
– Atomic mass spectrometry uses a mass spectrometer to separate the gaseous ions formed from the elements making up a sample of matter.
– The concentration of the resulting ions is then determined by measuring the electrical current produced when they fall on the surface of an ion detector.
What is Precipitation Gravimetry?
– In precipitation gravimetry, the analyte is converted to a sparingly soluble precipitate.
– This precipitate is then filtered, washed free of impurities, converted to a product of known composition by suitable heat treatment, and weighed.
– For example, a precipitation method for determining calcium in water is one of the official methods of the Association of Official Analytical Chemists.
– In this technique, an excess of oxalic acid, H2C2O4, is added to an aqueous solution of the sample.
– Ammonia is then added, which neutralizes the acid and causes essentially all of the calcium in the sample to precipitate as calcium oxalate.
– The reactions are:
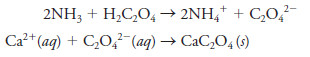
– The CaC2O4 precipitate is filtered using a weighed filtering crucible, then dried and ignited. This process converts the precipitate entirely to calcium oxide.
– The reaction is:
![]()
– After cooling, the crucible and precipitate are weighed, and the mass of calcium oxide is determined by subtracting the known mass of the crucible.
Properties of Precipitates and Precipitating Reagents
– Ideally, a gravimetric precipitating agent should react specifically or at least selectively with the analyte.
– Specific reagents, which are rare, react only with a single chemical species. Selective reagents, which are more common, react with a limited number of species.
– In addition to specificity and selectivity, the ideal precipitating reagent would react with the analyte to give a product that is
(1) easily filtered and washed free of contaminants;
(2) of sufficiently low solubility that no significant loss of the analyte occurs during filtration and washing;
(3) unreactive with constituents of the atmosphere;
(4) of known chemical composition after it is dried or, if necessary, ignited.
– Few, if any, reagents produce precipitates that have all these desirable properties.
Particle Size and Filterability of Precipitates
– Precipitates consisting of large particles are generally desirable for gravimetric work because these particles are easy to filter and wash free of impurities.
– In addition, precipitates of this type are usually purer than are precipitates made up of fine particles.
Factors That Determine the Particle Size of Precipitates
– The particle size of solids formed by precipitation varies enormously.
– At one extreme are colloidal suspensions, whose tiny particles are invisible to the naked eye (10-7 to 10-4 cm in diameter).
– Colloidal particles show no tendency to settle from solution and are difficult to filter.
– At the other extreme are particles with dimensions on the order of tenths of a millimeter or greater.
– The temporary dispersion of such particles in the liquid phase is called a crystalline suspension.
– The particles of a crystalline suspension tend to settle spontaneously and are easily filtered.
– Precipitate formation has been studied for many years, but the mechanism of the process is still not fully understood.
– What is certain, however, is that the particle size of a precipitate is influenced by precipitate solubility, temperature, reactant concentrations, and the rate at which reactants are mixed.
– The net effect of these variables can be accounted for, at least qualitatively, by assuming that the particle size is related to a single property of the system called relative supersaturation, where :
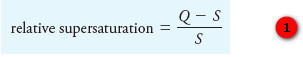
– Equation (1) is known as the Von Weimarn equation in recognition of the scientist who proposed it in 1925.
– In this equation, Q is the concentration of the solute at any instant, and S is its equilibrium solubility.
– Generally, precipitation reactions are slow so that, even when a precipitating reagent is added drop by drop to a solution of an analyte, some supersaturation is likely.
– Experimental evidence indicates that the particle size of a precipitate varies inversely with the average relative supersaturation during the time when the reagent is being introduced.
– Thus, when (Q – S)/S is large, the precipitate tends to be colloidal, and when (Q – S)/S is small, a crystalline solid is more likely.
Mechanism of Precipitate Formation
– The effect of relative supersaturation on particle size can be explained if we assume that precipitates form in two ways: by nucleation and by particle growth.
– The particle size of a freshly formed precipitate is determined by the mechanism that predominates.
In nucleation, a few ions, atoms, or molecules (perhaps as few as four or five) come together to form a stable solid.
– Often, these nuclei form on the surface of suspended solid contaminants, such as dust particles.
– Further precipitation then is governed by the competition between additional nucleation and growth of existing nuclei (particle growth).
– If nucleation predominates, a precipitate containing a large number of small particles results, and if growth predominates, a smaller number of larger particles is produced.
– The rate of nucleation is believed to increase enormously with increasing relative supersaturation.
– In contrast, the rate of particle growth is only moderately enhanced by high relative supersaturations.
– Therefore, when a precipitate is formed at high relative supersaturation, nucleation is the major precipitation mechanism, and a large number of small particles is formed.
– At low relative supersaturations, on the other hand, the rate of particle growth tends to predominate, and deposition of solid on existing particles occurs rather than further nucleation.
– Low relative supersaturation produces crystalline suspensions.
Experimental Control of Particle Size
– Experimental variables that minimize supersaturation and thus produce crystalline precipitates include elevated temperatures to increase the solubility of the precipitate (S in Equation (1)), dilute solutions (to minimize Q), and slow addition of the precipitating agent with good stirring.
– The last two measures also minimize the concentration of the solute (Q) at any given instant.
– If the solubility of the precipitate depends on pH, larger particles can also be produced by controlling pH.
– For example, large, easily filtered crystals of calcium oxalate are obtained by forming the bulk of the precipitate in a mildly acidic environment in which the salt is moderately soluble.
– The precipitation is then completed by slowly adding aqueous ammonia until the acidity is sufficiently low for removal of substantially all of the calcium oxalate.
– The additional precipitate produced during this step deposits on the solid particles formed in the first step.
– Unfortunately, many precipitates cannot be formed as crystals under practical laboratory conditions.
– A colloidal solid is generally formed when a precipitate has such a low solubility that S in Equation(1) always remains negligible relative to Q.
– The relative supersaturation thus remains enormous throughout precipitate formation, and a colloidal suspension results.
– For example, under conditions feasible for an analysis, the hydrous oxides of iron(III), aluminum, and chromium(III) and the sulfides of most heavy-metal ions form only as colloids because of their very low solubilities.
Colloidal Precipitates
– Individual colloidal particles are so small that they are not retained by ordinary filters. Moreover, Brownian motion prevents their settling out of solution under the influence of gravity.
– Fortunately, however, we can coagulate, or agglomerate, the individual particles of most colloids to give a filterable, amorphous mass that will settle out of solution.
Coagulation of Colloids
– Coagulation can be hastened by heating, by stirring, and by adding an electrolyte to the medium.
– To understand the effectiveness of these measures, we need to look into why colloidal suspensions are stable and do not coagulate spontaneously.
– Colloidal suspensions are stable because all of the particles of the colloid are either positively or negatively charged and thus repel one another.
– The charge results from cations or anions that are bound to the surface of the particles.
– We can show that colloidal particles are charged by placing them between charged plates where some of the particles migrate toward one electrode while others move toward the electrode of the opposite charge.
– The process by which ions are retained on the surface of a solid is known as adsorption.
– The adsorption of ions on an ionic solid originates from the normal bonding forces that are responsible for crystal growth.
– For example, a silver ion at the surface of a silver chloride particle has a partially unsatisfied bonding capacity for anions because of its surface location.
– Negative ions are attracted to this site by the same forces that hold chloride ions in the silver chloride lattice.
– Chloride ions at the surface of the solid exert an analogous attraction for cations dissolved in the solvent.
The kind of ions retained on the surface of a colloidal particle and their number depend in a complex way on several variables.
– For a suspension produced in a gravimetric analysis, however, the species adsorbed, and hence the charge on the particles, can be easily predicted because lattice ions are generally more strongly held than others.
– For example, when silver nitrate is first added to a solution containing chloride ion, the colloidal particles of the precipitate are negatively charged as a result of adsorption of some of the excess chloride ions.
– This charge, though, becomes positive when enough silver nitrate has been added to provide an excess of silver ions.
– The surface charge is at a minimum when the supernatant liquid does not contain an excess of either ion.
– The extent of adsorption and thus the charge on a given particle increase rapidly as the concentration of a common ion becomes greater.
– Eventually, however, the surface of the particles becomes covered with the adsorbed ions, and the charge becomes constant and independent of concentration.
– Figure (1) shows a colloidal silver chloride particle in a solution that contains an excess of silver nitrate.
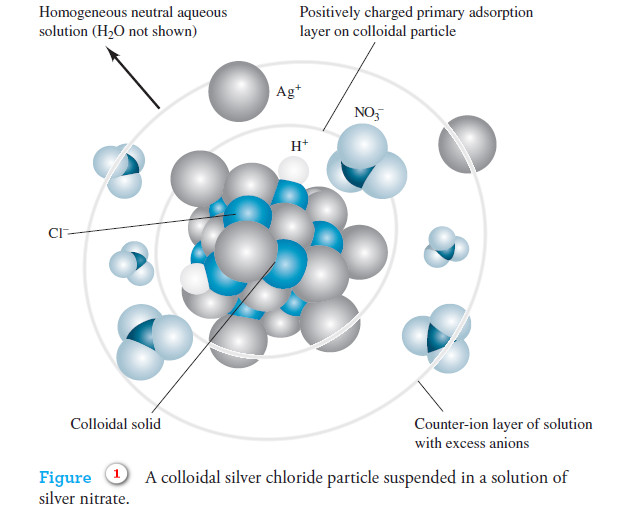
– Attached directly to the solid surface is the primary adsorption layer, which consists mainly of adsorbed silver ions.
– Surrounding the charged particle is a layer of solution, called the counter-ion layer, which contains sufficient excess of negative ions (principally nitrate) to just balance the charge on the surface of the particle.
– The primarily adsorbed silver ions and the negative counter-ion layer constitute an electric double layer that imparts stability to the colloidal suspension.
– As colloidal particles approach one another, this double layer exerts an electrostatic repulsive force that prevents particles from colliding and adhering.
– Figure (2a) shows the effective charge on two silver chloride particles.
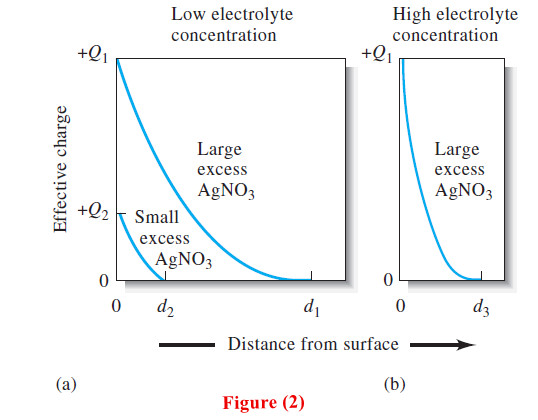
– The upper curve represents a particle in a solution that contains a reasonably large excess of silver nitrate, and the lower curve depicts a particle in a solution that has a much lower silver nitrate content.
– The effective charge can be thought of as a measure of the repulsive force that the particle exerts on like particles in the solution.
– Note that the effective charge falls off rapidly as the distance from the surface increases, and it approaches zero at the points d1 or d2.
– These decreases in effective charge (in both cases positive) are caused by the negative charge of the excess counter-ions in the double layer surrounding each particle.
– At points d1 and d2, the number of counterions in the layer is approximately equal to the number of primarily adsorbed ions on the surfaces of the particles; therefore, the effective charge of the particles approaches zero at this point.
– The upper portion of Figure (3) depicts two silver chloride particles and their counter- ion layers as they approach each other in the concentrated silver nitrate just considered.
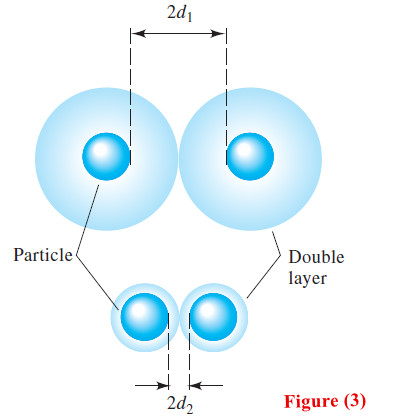
– Note that the effective charge on the particles prevents them from approaching one another more closely than about 2d1—a distance that is too great for coagulation to occur.
– As shown in the lower part of Figure (3), in the more dilute silver nitrate solution, the two particles can approach within 2d2 of one another.
– Ultimately, as the concentration of silver nitrate is further decreased, the distance between particles becomes small enough for the forces of agglomeration to take effect and a coagulated precipitate to appear.
– Coagulation of a colloidal suspension can often be brought about by a short period of heating, particularly if accompanied by stirring.
– Heating decreases the number of adsorbed ions and thus the thickness, di, of the double layer.
– The particles may also gain enough kinetic energy at the higher temperature to overcome the barrier to close approach imposed by the double layer.
– An even more effective way to coagulate a colloid is to increase the electrolyte concentration of the solution.
– If we add a suitable ionic compound to a colloidal suspension, the concentration of counter-ions increases in the vicinity of each particle.
– As a result, the volume of solution that contains sufficient counter-ions to balance the charge of the primary adsorption layer decreases.
– The net effect of adding an electrolyte is thus a shrinkage of the counter-ion layer, as shown in Figure (2b).
– The particles can then approach one another more closely and agglomerate.
Peptization of Colloids
– Peptization is the process by which a coagulated colloid reverts to its original dispersed state.
– When a coagulated colloid is washed, some of the electrolyte responsible for its coagulation is leached from the internal liquid in contact with the solid particles.
– Removal of this electrolyte has the effect of increasing the volume of the counter-ion layer.
– The repulsive forces responsible for the original colloidal state are then reestablished, and particles detach themselves from the coagulated mass.
– The washings become cloudy as the freshly dispersed particles pass through the filter.
– We are thus faced with a dilemma in working with coagulated colloids.
– On the one hand, washing is needed to minimize contamination, but on the other, there is a risk of losses resulting from peptization if pure water is used.
– The problem is usually solved by washing the precipitate with a solution containing an electrolyte that volatilizes when the precipitate is dried or ignited.
– For example, silver chloride is usually washed with a dilute solution of nitric acid.
– While the precipitate no doubt becomes contaminated with acid, no harm is done, since the nitric acid is lost during the drying step.
Practical Treatment of Colloidal Precipitates
– Colloids are best precipitated from hot, stirred solutions containing sufficient electrolyte to ensure coagulation.
– The filterability of a coagulated colloid often improves if it is allowed to stand for an hour or more in contact with the hot solution from which it was formed.
– During this process, which is known as digestion, weakly bound water appears to be lost from the precipitate. The result is a denser mass that is easier to filter.
Crystalline Precipitates
– Crystalline precipitates are generally more easily filtered and purified than are coagulated colloids.
– In addition, the size of individual crystalline particles, and thus their filterability, can be controlled to some extent.
Methods of Improving Particle Size and Filterability
– The particle size of crystalline solids can often be improved significantly by minimizing Q or maximizing S, or both, in Equation (1).
– The value of Q is can often be minimized by using dilute solutions and adding the precipitating reagent slowly, with good mixing.
– Often, S is increased by precipitating from hot solution or by adjusting the pH of the precipitation medium.
– Digestion of crystalline precipitates (without stirring) for some time after formation often yields a purer, more filterable product.
– The improvement in filterability undoubtedly results from the dissolution and recrystallization that occur continuously and at an enhanced rate at elevated temperatures.
– Recrystallization apparently results in bridging between adjacent particles, a process that yields larger and more easily filtered crystalline aggregates.
– This view is supported by the observation that little improvement in filtering characteristics occurs if the mixture is stirred during digestion.
Coprecipitation
– Coprecipitation is a process in which normally soluble compounds are carried out of solution by a precipitate
– When otherwise soluble compounds are removed from solution during precipitate formation, we refer to the process as coprecipitation.
– Contamination of a precipitate by a second substance whose solubility product has been exceeded is not coprecipitation.
– There are four types of coprecipitation: surface adsorption, mixed-crystal formation, occlusion, and mechanical entrapment.
– Surface adsorption and mixed-crystal formation are equilibrium processes, and occlusion and mechanical entrapment arise from the kinetics of crystal growth.
Surface Adsorption
Adsorption is a common source of coprecipitation and is likely to cause significant contamination of precipitates with large specific surface areas, that is, coagulated colloids.
– Although adsorption does occur in crystalline solids, its effects on purity are usually undetectable because of the relatively small specific surface area of these solids.
– Coagulation of a colloid does not significantly decrease the amount of adsorption because the coagulated solid still contains large internal surface areas that remain exposed to the solvent (Figure 4).
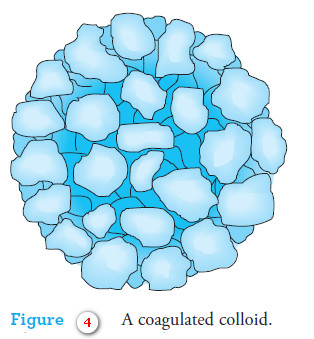
– The coprecipitated contaminant on the coagulated colloid consists of the lattice ion originally adsorbed on the surface before coagulation plus the counter-ion of opposite charge held in the film of solution immediately adjacent to the particle.
– The net effect of surface adsorption is, therefore, the carrying down of an otherwise soluble compound as a surface contaminant.
– For example, the coagulated silver chloride formed in the gravimetric determination of chloride ion is contaminated with primarily adsorbed silver ions with nitrate or other anions in the counter-ion layer.
– The result is that silver nitrate, a normally soluble compound, is coprecipitated with the silver chloride.
Minimizing Adsorbed Impurities on Colloids
– The purity of many coagulated colloids is improved by digestion.
– During this process, water is expelled from the solid to give a denser mass that has a smaller specific surface area for adsorption.
– Washing a coagulated colloid with a solution containing a volatile electrolyte may also be helpful because any nonvolatile electrolyte added earlier to cause coagulation is displaced by the volatile species.
– Washing generally does not remove much of the primarily adsorbed ions because the attraction between these ions and the surface of the solid is too strong.
– Exchange occurs, however, between existing counter-ions and ions in the wash liquid.
– For example, in the determination of silver by precipitation with chloride ion, the primarily adsorbed species is chloride. Washing with an acidic solution converts the counter-ion layer largely to hydrogen ions so that both chloride and hydrogen ions are retained by the solid. Volatile HCl is then given off when the precipitate is dried.
– Regardless of the method of treatment, a coagulated colloid is always contaminated to some degree, even after extensive washing.
– The error introduced into the analysis from this source can be as low as 1 to 2 ppt, as in the coprecipitation of silver nitrate on silver chloride.
– In contrast, coprecipitation of heavy-metal hydroxides on the hydrous oxides of trivalent iron or aluminum can result in errors as large as several percent, which is intolerable.
Reprecipitation
– A drastic but effective way to minimize the effects of adsorption is reprecipitation. In this process, the filtered solid is redissolved and reprecipitated.
– The first precipitate usually carries down only a fraction of the contaminant present in the original solvent.
– Thus, the solution containing the redissolved precipitate has a significantly lower contaminant concentration than the original, and even less adsorption occurs during the second precipitation.
– Reprecipitation adds substantially to the time required for an analysis. However, it is often necessary for such precipitates as the hydrous oxides of iron(III) and aluminum, which have extraordinary tendencies to adsorb the hydroxides of heavy-metal cations such as zinc, cadmium, and manganese.
Mixed-Crystal Formation
– Mixed-crystal formation is a type of coprecipitation in which a contaminant ion replaces an ion in the lattice of a crystal.
– In mixed-crystal formation, one of the ions in the crystal lattice of a solid is replaced by an
ion of another element.
– For this exchange to occur, it is necessary that the two ions have the same charge and that their sizes differ by no more than about 5%. Furthermore, the two salts must belong to the same crystal class.
– For example, barium sulfate formed by adding barium chloride to a solution containing sulfate, lead, and acetate ions is found to be severely contaminated by lead sulfate.
– This contamination occurs even though acetate ions normally prevent precipitation of lead sulfate by complexing the lead. In this case, lead ions replace some of the barium ions in the barium sulfate crystals.
– Other examples of coprecipitation by mixed-crystal formation include MgKPO4 in MgNH4PO4, SrSO4 in BaSO4, and MnS in CdS.
– The extent of mixed-crystal contamination is governed by the law of mass action and increases as the ratio of contaminant to analyte concentration increases.
– Mixedcrystal formation is a particularly troublesome type of coprecipitation because little can be done about it when certain combinations of ions are present in a sample matrix. This problem occurs with both colloidal suspensions and crystalline precipitates.
– When mixed-crystal formation occurs, the interfering ion may have to be separated before the final precipitation step.
– Alternatively, a different precipitating reagent that does not give mixed crystals with the ions in question may be used.
Occlusion and Mechanical Entrapment
– Occlusion is a type of coprecipitation in which a compound is trapped within a pocket formed during rapid crystal growth.
– When a crystal is growing rapidly during precipitate formation, foreign ions in the counter-ion layer may become trapped, or occluded, within the growing crystal.
– Because supersaturation and thus growth rate decrease as precipitation progresses, the amount of occluded material is greatest in that part of a crystal that forms first.
– Mechanical entrapment occurs when crystals lie close together during growth.
– Several crystals grow together and in so doing trap a portion of the solution in a tiny pocket.
– Both occlusion and mechanical entrapment are at a minimum when the rate of precipitate formation is low, that is, under conditions of low supersaturation.
– In addition, digestion often reduces the effects of these types of coprecipitation.
– Undoubtedly, the rapid dissolving and reprecipitation that occur at the elevated temperature of digestion open up the pockets and allow the impurities to escape into the solution.
Coprecipitation Errors
– Coprecipitated impurities may cause either negative or positive errors in an analysis.
– If the contaminant is not a compound of the ion being determined, a positive error will always result.
– Therefore, a positive error is observed whenever colloidal silver chloride adsorbs silver nitrate during a chloride analysis.
– In contrast, when the contaminant does contain the ion being determined, either positive or negative errors may occur.
– For example, in the determination of barium by precipitation as barium sulfate, occlusion of other barium salts occurs.
– If the occluded contaminant is barium nitrate, a positive error is observed because this compound has a larger molar mass than the barium sulfate that would have formed had no coprecipitation occurred.
– If barium chloride is the contaminant, the error is negative because its molar mass is less than that of the sulfate salt.
Precipitation from Homogeneous Solution
– Precipitation from homogeneous solution is a technique in which a precipitating agent is generated in a solution of the analyte by a slow chemical reaction.
– Local reagent excesses do not occur because the precipitating agent appears gradually and homogeneously throughout the solution and reacts immediately with the analyte.
– As a result, the relative supersaturation is kept low during the entire precipitation.
– In general, homogeneously formed precipitates, both colloidal and crystalline, are better suited for analysis than a solid formed by direct addition of a precipitating reagent.
– Urea is often used for the homogeneous generation of hydroxide ion. The reaction can be expressed by the equation:
![]()
– This hydrolysis proceeds slowly at temperatures just below 100°C, with 1 to 2 hours needed to complete a typical precipitation.
– Urea is particularly valuable for the precipitation of hydrous oxides or basic salts.
– For example, hydrous oxides of iron(III) and aluminum, formed by direct addition of base, are bulky and gelatinous masses that are heavily contaminated and difficult to filter.
– In contrast, when these same products are produced by homogeneous generation of hydroxide ion, they are dense, are easily filtered, and have considerably higher purity.
– Figure (5) shows hydrous oxide precipitates of aluminum formed by direct addition of base and by homogeneous precipitation with urea.

– Homogeneous precipitation of crystalline precipitates also results in marked increases in crystal size as well as improvements in purity.
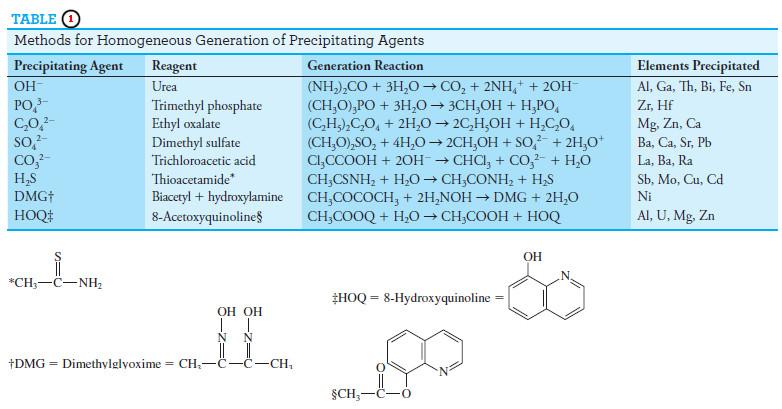
– Representative methods based on precipitation by homogeneously generated
reagents are given in Table (1).
Drying and Ignition of Precipitates
– After filtration, a gravimetric precipitate is heated until its mass becomes constant.
– Heating removes the solvent and any volatile species carried down with the precipitate.
– Some precipitates are also ignited to decompose the solid and form a compound of known composition. This new compound is often called the weighing form.
– The temperature required to produce a suitable weighing form varies from precipitate
to precipitate.
– Figure (6) shows mass loss as a function of temperature for several common analytical precipitates.

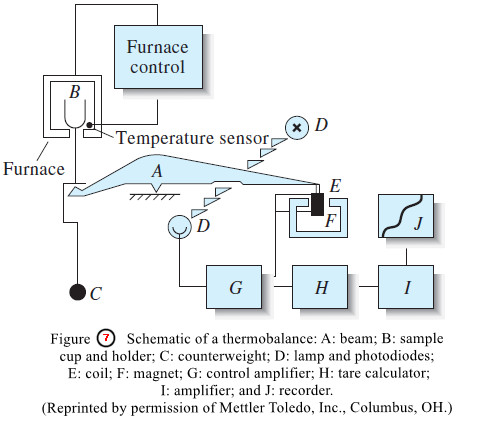
– These data were obtained with an automatic thermobalance, an instrument that records the mass of a substance continuously as its temperature is increased at a constant rate (Figure 7).
– Heating three of the precipitates— silver chloride, barium sulfate, and aluminum oxide—simply causes removal of water and perhaps volatile electrolytes.
– Note the vastly different temperatures required to produce an anhydrous precipitate of constant mass.
– Moisture is completely removed from silver chloride at temperatures higher than 110°C, but dehydration of aluminum oxide is not complete until a temperature greater than 1000°C is achieved.
– Aluminum oxide formed homogeneously with urea can be completely dehydrated at about 650°C.
– The thermal curve for calcium oxalate is considerably more complex than the others shown in Figure (6).
– Below about 135°C, unbound water is eliminated to give the monohydrate CaC2O4.H2O. This compound is then converted to the anhydrous oxalate CaC2O4 at 225°C.
– The abrupt change in mass at about 450°C signals the decomposition of calcium oxalate to calcium carbonate and carbon monoxide.
– The final step in the curve depicts the conversion of the carbonate to calcium oxide and carbon dioxide.
– As can be seen, the compound finally weighed in a gravimetric calcium determination based on oxalate precipitation is highly dependent on the ignition temperature.
Reference
- Modern analytical chemistry / David Harvey / The McGraw-Hill Companies, Inc./ , 2000 . USA
- Dean’s Analytical Chemistry Handbook / Pradyot Patnaik / The McGraw-Hill Companies, 2nd Editionm, 2004 .USA
- Fundamentals of analytical chemistry / Douglas A. Skoog, Donald M. West, F. James Holler, Stanley R. Crouch. (ninth edition) , 2014 . USA





